
AmBeed文献解读|JACS
给电子基团和吸电子基团对羰基化合物电化学氢解和氢化的影响

2024 年 5 月 21 日,美国威斯康星大学麦迪逊分校的J. R. Schmidt课题组和Kyoung-Shin Choi课题组在JACS发表题为《The Impact of Electron Donating and Withdrawing Groups on Electrochemical
Hydrogenolysis and Hydrogenation of Carbonyl Compounds》的研究。研究结果表明较强的供电子基团促进氢解,而较强的吸电子基团促进氢化。这项研究提供了对反应途径中涉及的中间体、过渡态和动力学势垒的深入理解,并且为控制羰基化合物的氢解/氢化选择性提供了关键信息和见解。
研究背景
羰基的氢解或氢化反应,即将C=O基团转化为CH2基团,在许多领域具有重要意义。在电化学实现羰基氢解过程中,如何以高选择性实现氢解存在挑战,因为羰基的电化学氢化,即将C=O基团转化为醇基团(CH-OH),并不是氢解的初始步骤。相反,氢化和氢解是同时发生的,并且它们是竞争性反应。这意味着尽管氢解和氢化都需要向羰基基团添加氢原子,但它们涉及的是在电极表面形成的不同中间体。因此,揭示氢解和氢化途径中的中间体、过渡态和动力学势垒的差异是理解和控制羰基化合物氢解/氢化选择性的关键。
研究内容
本课题旨在研究分子本身影响其氢解/氢化选择性的特征。作者研究了在锌阴极上具有不同供电子/吸电子能力的对位取代苯甲醛化合物的电化学还原反应,反应物选择范围及相应的Hammett常数如下所示(图 1)。
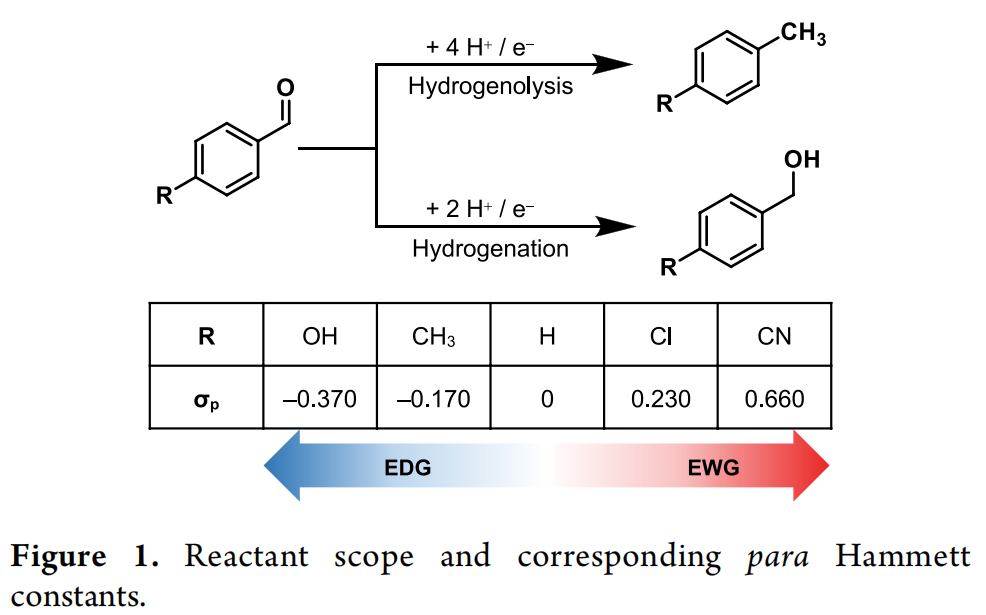
图 1. 反应物范围和相应的苯甲醛对位取代化合物的Hammett常数
作者首先使用锌金属工作电极,在有无10 mM的醛类反应物溶液中记录线性扫描伏安图(LSVs)。醛类反应物的加入使还原的起始电位向正向移动300-500 mV,表明本研究中使用的所有醛都比锌的析氢反应(HER)更容易。此外,所有醛类化合物的LSVs,在醛还原的起始电位和析氢反应(HER)的起始电位之间都清楚地显示出电流增强,这表明在这个电位区域内有可能实现具有高的法拉第效率(FE)的有机还原反应。然而,LSVs没有提供任何关于醛还原反应(即氢解和氢化)与还原电流有关的信息。
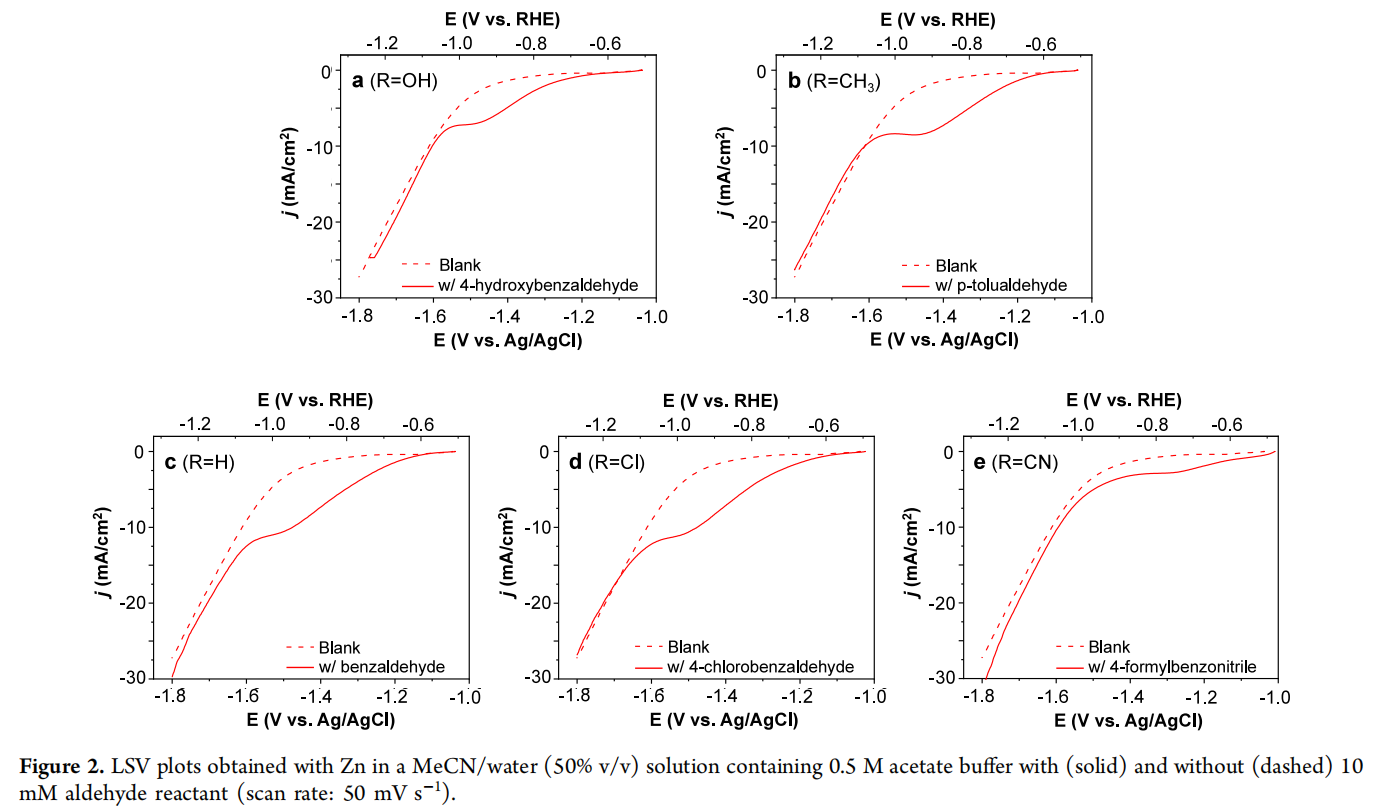
图 2. 在含有0.5 M醋酸缓冲液的MeCN/水(50% v/v)溶液中,分别在有(实线)和无(虚线)10 mM醛类反应物的情况下(扫描速率:50 mV/s),使用锌(Zn)得到的线性扫描伏安图(LSVs)
接下来,作者在-1.46 V的参比电极Ag/AgCl下进行了恒电位电解。结果显示,供电子基团(EDGs)和吸电子基团(EWGs)对醛基的氢解和氢化有显著的效应:EDGs促进氢解,而EWGs促进氢化。例如,随着取代基的吸电子能力增强,氢解产物的浓度从3.23 ± 0.09 mM(R = OH)下降到0.02 ± 0.0003 mM(R = CN)(图 3a),氢解的FE从64.6% ± 1.9%(R = OH)下降到0.44% ± 0.01%(R = CN)(图 3b)。当取代基的吸电子能力增强时,氢解的FE下降幅度比氢化FE的上升幅度要大,导致氢解和氢化的组合FE在取代基系列的中间位置出现下降。在相对选择性实验中,EDGs和EWGs对氢解和氢化的影响也很明显,这里只考虑了醛基氢解和氢化的选择性,它们的总和为100%(图 3c)。
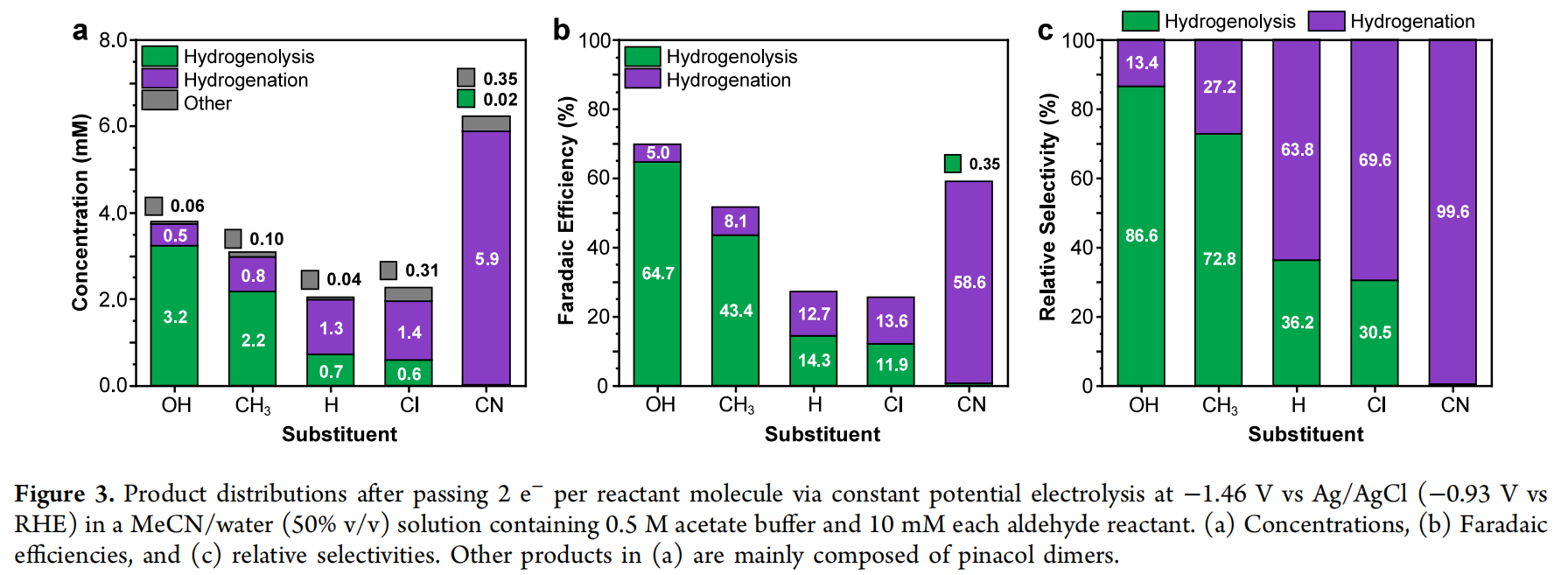
图 3. 通过在-1.46 V的Ag/AgCl(相对于RHE的-0.93 V)下恒电位电解,在含有0.5 M醋酸缓冲液和10 mM每种醛类反应物的MeCN/水(体积比50% v/v)溶液中,每分子反应物传递2e-后的产物分布图。(a) 浓度,(b) 法拉第效率,(c) 相对选择性。在(a)中的其他产物主要由频哪醇二聚体组成。
随着取代基吸电子能力的增强,氢解的偏电流密度减小(从4.57 ± 0.21降低到0.02 ± 0.0003 mA/cm2)。另一方面,随着取代基吸电子能力的增强,氢化的偏电流密度增加(从0.35 ± 0.04增加到2.94 ± 0.01 mA/cm2)。根据Hammett常数绘制的氢解和氢化的FE,显示出氢解和氢化的相同的系统和相反的趋势(图 4c,d)。EDGs和EWGs以相反的方式影响氢解和氢化选择性,暗示了氢解和氢化途径中,关键竞争步骤所涉及的过渡态的电荷可能具有相反特性。
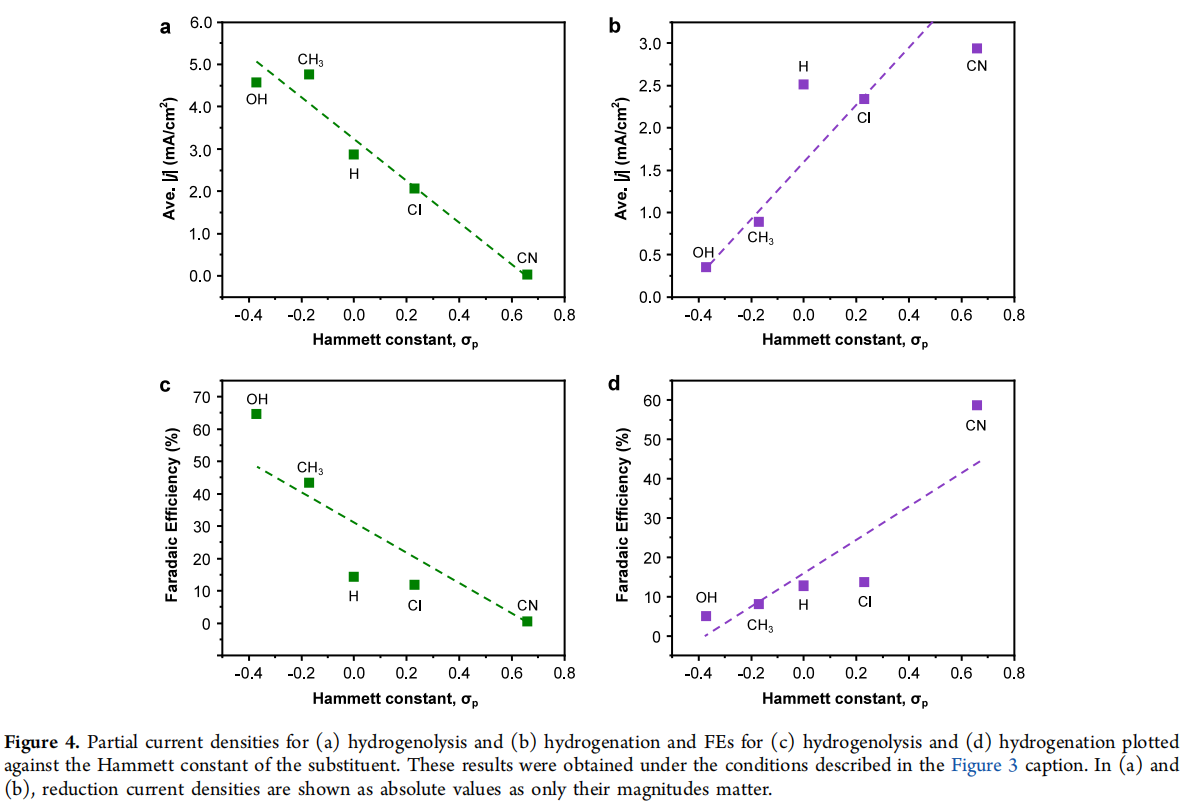
图 4. (a)氢解的偏电流密度和(b)氢化的偏电流密度,以及(c)氢解的法拉第效率(FEs)和(d)氢化的法拉第效率(FEs),与取代基的Hammett常数对比图。
计算研究表明,OH:CHO*、H:CHO*和Cl:CHO*在整个电位范围内(从−0.8到−1.6 V vs SHE)与Zn表面没有特定化学吸附或共价键形成的迹象。而CN:CHO*分子在电位比−1.2 V(相对于SHE)更低时,与Zn表面形成化学键,显示出吸附几何结构和能量的显著变化。作者比较了不同取代基的还原中间体的自由能和质子耦合电子转移(PCET)动力学势垒。结果显示,取代基的供电子/吸电子能力显著影响CHOH*到CH2OH*的PCET氢化路径的动力学势垒。吸电子基团能够稳定过渡态,降低反应势垒,从而增加氢化的选择性。对于氢解的关键步骤(将CHOH*转化为CH*),其过渡态并未显著受到取代基性质的影响。然而,由于氢化途径的过渡态受到取代基影响,这直接与氢解途径竞争,因此氢解的偏电流密度和法拉第效率受到了取代基的供电子/吸电子能力的影响。计算研究提供了对氢解和氢化途径中涉及的关键过渡态的深入理解,有助于揭示EDGs和EWGs如何影响这些途径的动力学和热力学特性。
结论
研究表明EDGs(供电子基团)和EWGs(吸电子基团)可以系统地影响苯甲醛在Zn上的电化学氢解/氢化的选择性。较强的EDG促进了羰基的氢解,而较强的EWG促进了羰基的氢化。计算研究表明,关键氢化步骤(将CHOH*转化为CH2OH*)的过渡态涉及在羰基碳原子上形成一个自由基,这可以通过吸电子基团达到稳定效果。因此,随着取代基的吸电子能力的增强,这一步骤的PCET动力学势垒降低。另一方面,关键氢解步骤(将CHOH*转化为CH*)的过渡态并未显著受到取代基的影响。然而,由于氢化途径的过渡态受到取代基影响,这直接与氢解途径竞争,氢解的偏电流密度和法拉第效率受到了取代基的供电子/吸电子能力的影响。吸电子能力最强的CN基团导致苯甲醛分子的吸附几何结构倾斜,形成了Zn-羰基键,这采用了一条新的PCET氢化途径(通过形成CH2O*),对于其他取代基的苯甲醛来说,这在动力学上是不可行的。由于CN基团有两种不同的促进氢化的方式,在CN基团存在下,苯甲醛的氢解几乎是不可能的。此外,作者发现将CHO*转化为CH2O*的HAT(氢原子转移)动力学势垒,随着取代基的吸电子能力的增强而降低,只能产生氢化产物。因此,尽管预计Zn上的稳态H*覆盖率不会很高,在PCET速率较慢的高pH溶液中,通过HAT的氢化不可忽视,其对氢化选择性的贡献随着取代基的吸电子能力而增加。本课题为研究关键氢解和氢化步骤的过渡态提供了重要信息,推进了电化学克莱门森还原的理解。

图 5. 实验所用的4-(hydroxymethyl)benzonitrile (874-89-5, A295046) 来自AmBeed品牌
文献DOI: http://10.1021/jacs.4c03032