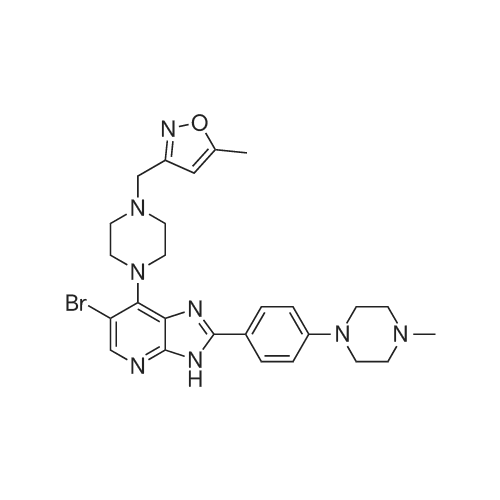
CCT 137690
产品说明书

 |
同义名 : | - |
| CAS号 : | 1095382-05-0 | |
| 货号 : | A917251 | |
| 分子式 : | C26H31BrN8O | |
| 纯度 : | 99%+ | |
| 分子量 : | 551.481 | |
| MDL号 : | MFCD18206879 | |
| 存储条件: |
Pure form Keep in dark place,Inert atmosphere,2-8°C In solvent -20°C:3-6个月-80°C:12个月 |
|
| 溶解度 : |
DMSO: 16 mg/mL(29.01 mM),配合低频超声助溶,注意:DMSO长时间开封后,会吸水并导致溶解能力下降,请避免使用长期开封的DMSO |
|
| 动物实验配方: |
1% DMSO+30% polyethylene glycol+1% Tween 80+water 30 mg/mL suspension |
| 生物活性 | |||
|---|---|---|---|
| 靶点 |
|
||
| 描述 | Aurora kinases are a family of 3 highly homologous serine/threonine kinases, and include 3 major members, AURKA, AURKB, and AURKC, that play a well-known role in the regulation of cell cycle and division. CCT137690, a pan-Aurora inhibitor, inhibits the proliferation of cancer cell lines of different origins including colorectal, ovarian, neuroblastoma and leukemia. CCT137690 is a highly selective, orally bioavailable imidazo[4,5-b]pyridine derivative that inhibits Aurora A and B kinases with low nanomolar IC50 values. The IC50 values are 15 nM, 25 nM, and 19 nM for aurora A, B and C, respectively. CCT137690 efficiently inhibits histone H3 and transforming acidic coiled-coil 3 phosphorylation (Aurora B and Aurora A substrates, respectively) in HCT116 and HeLa cells. Continuous exposure of tumor cells to CCT137690 causes multipolar spindle formation, chromosome misalignment, polyploidy, and apoptosis. The PDAC cell lines (PANC1, PANC2.03, CFPAC1, MiaPaCa2, BxPc3, and PANC02) and normal HPDEs cell line were used to identify CCT137690 with anticancer activity against PDAC ( pancreatic ductal adenocarcinoma cancer). Cells were treated with CCT137690 range from 0 - 40 μM for 24 hours, and the PDAC cells viability were significantly decreased by a dose depend manner. In contrast, normal HPDEs were resistant to CCT137690 treatment. Colony formation assays confirmed that the reproductive integrity of the PDAC cells after CCT137690 treatment was significantly reduced. Altogether, these results suggest that CCT137690 has anticancer activity in human PDAC cells. Human PANC1 cells implanted mice were administered CCT137690 orally at 80 mg/kg consecutively for 40 days. Compared with the vehicle control group, administration of CCT137690 effectively reduced tumor growth. Western blot analysis of indicated protein expressions in isolated tumor at day 34 showed that CCT137690 reduced the local phosphorylation of AURKA and GSK3β. These data demonstrated that CCT137690 significantly suppressed PDAC growth in vitro and in vivo through induction of necroptosis[3]. | ||
| 作用机制 | CCT137690 binds with Aurora-A enzyme, and the pyridine N and imidazole NH are interacting with Ala213 in the hinge region of the kinase. It occupies the ATP-binding site with the activation loop in a DFG-in conformation. The pyridine N is hydrogen bonded to backbone NH of Ala213 and the imidazole NH to the carbonyl of Ala213[4]. | ||
| 实验方案 | |||
|---|---|---|---|
| 1mg | 5mg | 10mg | |
|
1 mM 5 mM 10 mM |
1.81mL 0.36mL 0.18mL |
9.07mL 1.81mL 0.91mL |
18.13mL 3.63mL 1.81mL |

| 参考文献 |
|---|