Ulixertinib
产品说明书

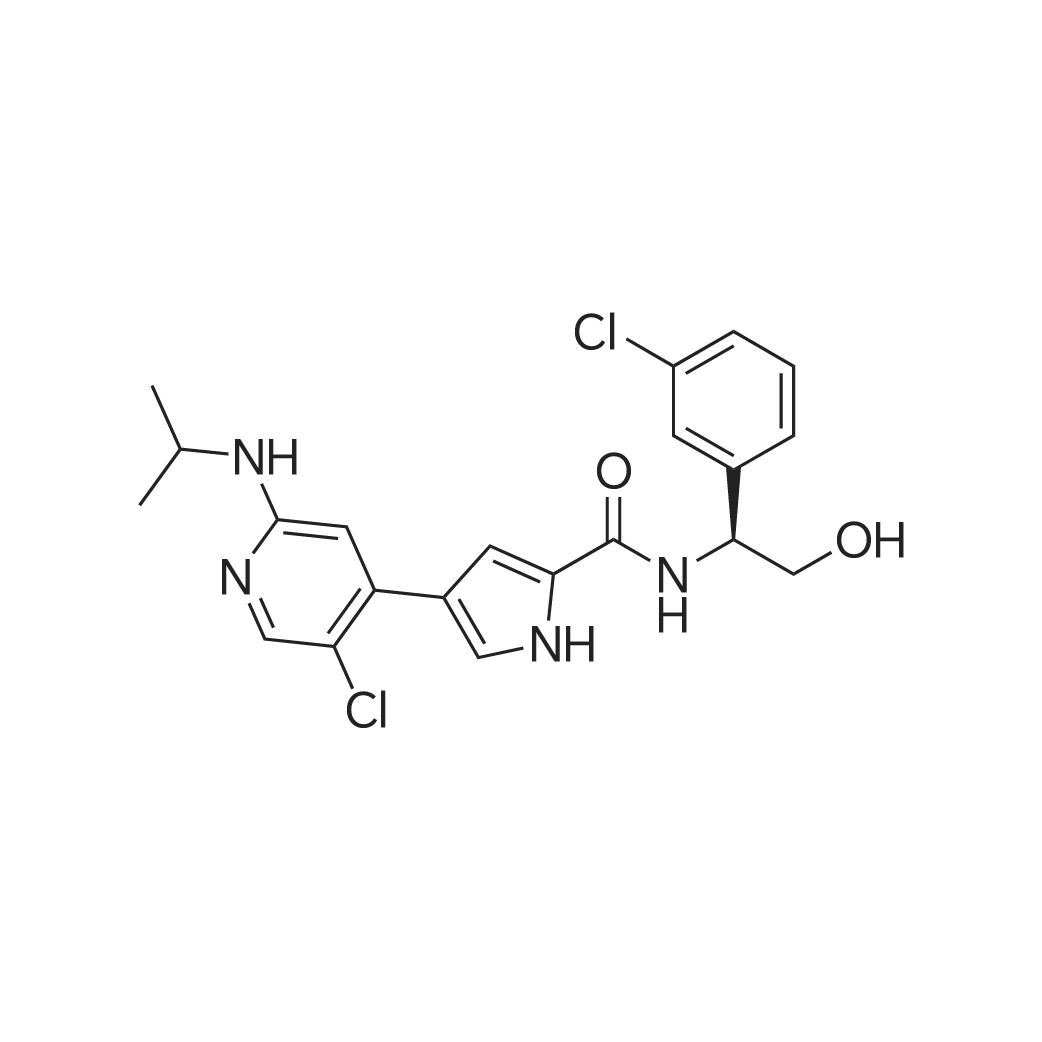 |
同义名 : | BVD-523;VRT752271 |
| CAS号 : | 869886-67-9 | |
| 货号 : | A162263 | |
| 分子式 : | C21H22Cl2N4O2 | |
| 纯度 : | 99%+ | |
| 分子量 : | 433.331 | |
| MDL号 : | MFCD22628898 | |
| 存储条件: |
Pure form Inert atmosphere,2-8°C In solvent -20°C:3-6个月-80°C:12个月 |
|
| 溶解度 : |
DMSO: 105 mg/mL(242.31 mM),配合低频超声助溶,注意:DMSO长时间开封后,会吸水并导致溶解能力下降,请避免使用长期开封的DMSO |
|
| 动物实验配方: |
IP 2% DMSO+2% Tween80+40% PEG300+water 8 mg/mL clear PO 0.5% CMC-Na 40 mg/mL suspension |
| 生物活性 | |||
|---|---|---|---|
| 靶点 |
|
||
| 描述 | ERK1/2 (extracellular-signal-regulated kinases) is only known substrates of the MEK1/2 as well as the essential node in RAS/RAF/MEK/ERK signaling cascade, which can be activated by catalyzing the phosphorylation on T202/Y204 through MEK1/2. Ulixertinib is a selective ERK1/2 inhibitor with IC50 value <0.3nM for ERK2, and Ki value of 0.3nM/0.04nM for ERK1/2[1][2]. The cellular study showed that Ulixertinib can inhibited p-ERK/p-RSK (the ERK substrate) with IC50 values of 4.1/0.14μM, as well as cell proliferation with IC50 value of 0.18μM of A375 cells[1]. Treatment with Ulixertinib at concentration of 2μM for 24h caused decrease of the ERK substrate, p-RSK, and the transcription targeted by ERK1/2, DUSP6, in BRAFV600E-mutant A375 melanoma cells. Meanwhile the increased p-ERK1/2 levels by this ATP-competitive inhibitor can also be observed. Ulixertinib at 0.25-4μM for 24h caused G1-arrested concentration-dependently in BRAFV600E-mutant melanoma cell line, UACC-62. A cell-line–dependent increase in caspase-3/7 by Ulixertinib was observed, as 3-fold greater induction of caspase-3/7 in a subset of cell lines with sensitivity to Ulixertinib (IC50<2μM). This could be further confirmed by attenuated p-RB and Cyclin D1, as well as the increased apoptotic marker BIM-EL by Ulixertinib in cells. Oral administration with Ulixertinib at 50 and 100mg/kg twice daily for 2 weeks suppressed tumor growth in A375 xenografts. The suppression of Colo205 tumors by Ulixertinib for 18 days could be observed at dose of 100mg/kg. Combination therapy with BVD-523 at dose of 100mg/kg with BRAF inhibitor dabrafenib (50mg/kg) can produce produced a 100% regression and a maximum 100% tumor growth delay in an A375 BRAFV600E-mutant melanoma model[2]. | ||
| 作用机制 | Ulixertinib is an ATP-competitive ERK1/2 inhibitor. Notice that measuring increased pERK1/2 levels could be considered as a pharmacodynamic biomarker for Ulixertinib, though inhibition of ERK1/2 targets such as pRSK or DUSP6 could still be observed.[2] | ||
| 细胞研究 | |||||
|---|---|---|---|---|---|
| 细胞系 | 浓度 | 检测类型 | 检测时间 | 活性说明 | 数据源 |
| human A375 cells | Function assay | 2 h | Inhibition of ERK1/2 in human A375 cells harboring B-RAF V600E mutant assessed as decrease in phospho-RSK level after 2 hrs by Cellomics ArrayScanTM VTI imaging analysis, IC50=0.14 μM | 25977981 | |
| human A375 cells | Proliferation assay | 72 h | Antiproliferative activity against human A375 cells harboring B-RAF V600E mutant after 72 hrs by Cellomics ArrayScanTM VTI imaging analysis, IC50=0.18 μM | 25977981 | |
| 实验方案 | |||
|---|---|---|---|
| 1mg | 5mg | 10mg | |
|
1 mM 5 mM 10 mM |
2.31mL 0.46mL 0.23mL |
11.54mL 2.31mL 1.15mL |
23.08mL 4.62mL 2.31mL |

| 参考文献 |
|---|